
Marfan syndrome
Aortic aneurysm - Marfan
Marfan syndrome is a disorder of connective tissue. This is the tissue that strengthens the body's structures.
Disorders of connective tissue affect the skeletal system, cardiovascular system, eyes, and skin.
Causes
Marfan syndrome is caused by variants in a gene called fibrillin-1. Fibrillin-1 plays an important role as the building block for connective tissue in the body.
The gene variant also causes the long bones of the body to grow too much. People with this syndrome have tall height and long arms and legs. How this overgrowth happens is not well understood.
Other areas of the body that are affected include:
- Lung tissue (there may be a pneumothorax, in which air can escape from the lung into the chest cavity and collapse the lung)
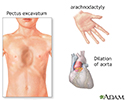
- The aorta, the main blood vessel that takes blood from the heart to the body may stretch or become weak (called aortic dilation or aortic aneurysm)
- The heart valves
- The eyes, causing cataracts and other problems (such as a dislocation of the lenses)
- The skin
- Tissue covering the spinal cord
- The joints
In most cases, Marfan syndrome is passed down through families (inherited). However, up to 30% of people have no family history, which is called "sporadic." In sporadic cases, the syndrome is believed to be caused by a new gene change.
Symptoms
People with Marfan syndrome are often tall with long, thin arms and legs and spider-like fingers (called arachnodactyly). The length of the arms is greater than height when arms are stretched out.
Other symptoms include:
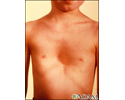
- A chest that sinks in or sticks out, called funnel chest (pectus excavatum) or pigeon breast (pectus carinatum)
- Flat feet
- Highly arched palate and crowded teeth
- Hypotonia of muscles (low muscle tone)
- Joints that are too flexible (but the elbows may be less flexible)
- Learning disability
- Movement of the lens of the eye from its normal position (dislocation)
- Nearsightedness
- Small lower jaw (micrognathia)
- Spine that curves to one side (scoliosis)
- Thin, narrow face




Many people with Marfan syndrome suffer from chronic muscle and joint pain.
Exams and Tests
The health care provider will perform a physical exam. The joints may move around more than normal. There may also be signs of:
- Aneurysm
- Collapsed lung
- Heart valve problems

An eye exam may show:
- Defects of the lens or cornea
- Retinal detachment
- Vision problems

The following tests may be performed:
- Echocardiogram
- Fibrillin-1 mutation testing (in some people)

An echocardiogram or another test should be done every year to look at the base of the aorta and possibly the heart valves. Depending on the results, you may need this test less often than yearly.
Treatment
Vision problems should be treated when possible.
Monitor for scoliosis, especially during the teenage years.
Medicine to slow the heart rate and lower blood pressure may help prevent stress on the aorta. To avoid injuring the aorta, people with the condition may have to modify their activities. Some people may need surgery to replace the aortic root and valve.
Pregnant women with Marfan syndrome must be monitored very closely because of the increased stress on the heart and aorta.
Support Groups
More information and support for people with Marfan syndrome and their families can be found at:
- The Marfan Foundation -- marfan.org
Outlook (Prognosis)
Heart-related complications may shorten the lifespan of people with this disease. However, many people live into their 60s and beyond. Good care and surgery may further extend lifespan.
Possible Complications
Complications may include:
- Aortic regurgitation
- Aortic rupture
- Bacterial endocarditis
- Dissecting aortic aneurysm (also called aortic dissection)
- Enlargement of the base of the aorta
- Heart failure
- Mitral valve prolapse
- Scoliosis
- Vision problems



When to Contact a Medical Professional
Couples who have this condition and are planning to have children may want to talk to a genetic counselor before starting a family.
Prevention
Spontaneous new gene variants leading to Marfan syndrome (less than one third of cases) cannot be prevented. If you have Marfan syndrome, see your provider at least once every year.
References
Doyle JJ, Dietz HC. Marfan syndrome. In: Kliegman RM, St. Geme JW, Blum NJ, et al. eds. Nelson Textbook of Pediatrics. 22nd ed. Philadelphia, PA: Elsevier; 2025:chap 743.
Madan-Khetarpal S, Arnold G, Ortiz D. Genetic disorders and dysmorphic conditions. In: Zitelli BJ, McIntire SC, Nowalk AJ, Garrison J, eds. Zitelli and Davis' Atlas of Pediatric Diagnosis. 8th ed. Philadelphia, PA: Elsevier; 2023:chap 1.
Pyeritz RE. Inherited diseases of connective tissue. In: Goldman L, Cooney KA, eds. Goldman-Cecil Medicine. 27th ed. Philadelphia, PA: Elsevier; 2024:chap 239.
Review Date: 5/8/2024

 All rights reserved.
All rights reserved.