
Thalassemia
Mediterranean anemia; Cooley anemia; Beta thalassemia; Alpha thalassemia
Thalassemia is a blood disorder passed down through families (inherited) in which the body makes an abnormal form or inadequate amount of hemoglobin. Hemoglobin is the protein in red blood cells that carries oxygen. The disorder results in large numbers of red blood cells being destroyed, which leads to anemia.
Causes
Hemoglobin is made of two proteins:
- Alpha globin
- Beta globin
Thalassemia occurs when there is a variant in a gene that helps control production of one of these proteins.
There are two main types of thalassemia:
- Alpha thalassemia occurs when a gene or genes related to the alpha globin protein are missing or there is a variant.
- Beta thalassemia occurs when similar gene defects affect production of the beta globin protein.
Alpha thalassemias occur most often in people from Southeast Asia, the Middle East, China, and in those of African descent.
Beta thalassemias occur most often in people of Mediterranean origin. To a lesser extent, Chinese, other Asians, and African Americans can be affected.
There are many forms of thalassemia. Each type has many different subtypes. Both alpha and beta thalassemia include the following two forms:
- Thalassemia major
- Thalassemia minor
You must inherit the variant gene from both parents to develop thalassemia major.
Thalassemia minor occurs if you receive the variant gene from only one parent. People with this form of the disorder are carriers of the disease. Most of the time, they do not have symptoms.
Beta thalassemia major is also called Cooley anemia.
Risk factors for thalassemia include:
- Asian, Chinese, Mediterranean, or African American ethnicity
- Family history of the disorder
Symptoms
The most severe form of alpha thalassemia major causes stillbirth (death of the unborn baby during birth or the late stages of pregnancy).
Children born with beta thalassemia major (Cooley anemia) are normal at birth, but develop severe anemia during the first year of life.

Other symptoms can include:
- Bone deformities in the face
- Fatigue
- Growth failure
- Shortness of breath
- Yellow skin (jaundice)

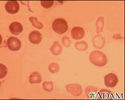
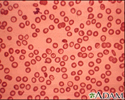
People with the minor form of alpha and beta thalassemia have small red blood cells but no symptoms.
Exams and Tests
Your health care provider will do a physical exam to look for an enlarged spleen.
A blood sample will be sent to a laboratory to be tested.
- Red blood cells will appear small and abnormally shaped when looked at under a microscope.
- A complete blood count (CBC) reveals anemia.
- A test called hemoglobin electrophoresis shows the presence of an abnormal form of hemoglobin and can detect beta thalassemia.
- A test called mutational analysis can help detect alpha thalassemia.

Treatment
Treatment for thalassemia major often involves regular blood transfusions and folate supplements.
If you receive blood transfusions, you should not take iron supplements. Doing so can cause a high amount of iron to build up in the body, which can be harmful.
People who receive a lot of blood transfusions need a treatment called chelation therapy. This is done to remove excess iron from the body.
A bone marrow transplant may help treat the disease in some people, especially children.
Outlook (Prognosis)
Severe thalassemia can cause early death (between ages 20 and 30) due to heart failure. Getting regular blood transfusions and therapy to remove iron from the body helps improve the outcome.
Less severe forms of thalassemia often do not shorten lifespan.
You may want to seek genetic counseling if you have a family history of the condition and are thinking of having children.
Possible Complications
Untreated, thalassemia major leads to heart failure and liver problems. It also makes a person more likely to develop infections.
Blood transfusions can help control some symptoms, but carry a risk of side effects from too much iron.
When to Contact a Medical Professional
Contact your provider if:
- You or your child has symptoms of thalassemia.
- You are being treated for the disorder and new symptoms develop.
References
Cappellini MD. The thalassemias. In: Goldman L, Cooney KA, eds. Goldman-Cecil Medicine. 27th ed. Philadelphia, PA: Elsevier; 2024:chap 148.
Owusu-Ansah A, Letterio J, Ahuja SP. Red blood cell disorders in the fetus and neonate. In: Martin RJ, Fanaroff AA, eds. Fanaroff and Martin's Neonatal-Perinatal Medicine. 12th ed. Philadelphia, PA: Elsevier; 2025:chap 81.
Sheth S.Thalassemia syndromes. In: Hoffman R, Benz EJ, Silberstein LE, et al, eds. Hematology: Basic Principles and Practice. 8th ed. Philadelphia, PA: Elsevier; 2023:chap 41.
Thom CS, Lambert MP. Anemia in the newborn infant. In: Kliegman RM, St. Geme JW, Blum NJ, et al, eds. Nelson Textbook of Pediatrics. 22nd ed. Philadelphia, PA: Elsevier; 2025:chap 139.
Review Date: 3/31/2024
Reviewed By: Todd Gersten, MD, Hematology/Oncology, Florida Cancer Specialists & Research Institute, Wellington, FL. Review provided by VeriMed Healthcare Network. Also reviewed by David C. Dugdale, MD, Medical Director, Brenda Conaway, Editorial Director, and the A.D.A.M. Editorial team.
 All rights reserved.
All rights reserved.