
Spinal muscular atrophy
Werdnig-Hoffmann disease; Kugelberg-Welander disease
Spinal muscular atrophy (SMA) is a group of disorders of the motor neurons (motor cells). These disorders are passed down through families (inherited) and can appear at any stage of life. The disorder leads to muscle weakness and atrophy.
Causes
SMA is a collection of different motor nerve (or neuron) diseases. The disease is caused by a lack of a protein (SMN) due to defective genes.
Most of the time, a person must get one copy of the defective gene from both parents to be affected. The most severe form is SMA type I, also called Werdnig-Hoffman disease. Infants with SMA type II have less severe symptoms during early infancy, but they become weaker with time. SMA type III is a less severe form of the disease.
In rare cases, SMA begins in adulthood. This is the mildest form of the disease.
A family history of SMA in an immediate family member (such as brother or sister) is a risk factor for all types of the disorder.
Symptoms
Symptoms of SMA may vary depending on the SMA type.
- Infants with SMA type I are born with very little muscle tone, weak muscles, and feeding and breathing problems.
- In infants with SMA type II, symptoms may not appear until age 6 months to 2 years.
- Type III SMA is a milder disease that starts in childhood or adolescence and slowly gets worse.
- Type IV is even milder, with weakness starting in adulthood.
Often, weakness is first felt in the shoulder and leg muscles. Weakness gets worse over time and eventually becomes severe.
Symptoms in an infant:
- Breathing difficulty with shortness of breath and labored breathing, leading to a lack of oxygen
- Feeding difficulty (food may go into the windpipe instead of the stomach)
- Floppy infant (poor muscle tone)
- Lack of head control
- Little movement
- Weakness that gets worse
Symptoms in a child:
- Frequent, increasingly severe respiratory infections
- Nasal speech
- Posture that gets worse
With SMA, the nerves that control feeling (sensory nerves) are not affected. So, a person with the disease can feel things normally.
Exams and Tests
The health care provider will take a careful history and perform a brain/nervous system (neurologic) examination to find out if there is:
- A family history of neuromuscular disease
- Floppy (flaccid) muscles
- No deep tendon reflexes
- Twitches of the tongue muscle
Tests that may be ordered include:
- Aldolase blood test
- Erythrocyte sedimentation rate (ESR)
- Creatine phosphate kinase blood test
- DNA testing to confirm diagnosis
- Electromyography (EMG)
- Lactate/pyruvate ratio in the blood
- MRI of the brain, spine, and spinal cord
- Muscle biopsy
- Nerve conduction study
- Amino acid blood tests
- Lung function tests
- Swallowing study
- Thyroid-stimulating hormone (TSH) blood test






Treatment
There is no treatment to cure SMA. However, there are now three drugs approved by the FDA that slow how fast the muscle weakness progresses:
- Onasemnogene abeparvovec-xioi (Zolgensma)
- Risdiplam (Evrysdi)
- Nusinersen (Spinraza)
These drugs work by increasing the amount of the SMN protein produced. Talk with your provider to see if either of these medicines is right for you or your child.
Supportive care is important. Breathing complications are common in the more severe forms of SMA. To help with breathing, a device or machine called a ventilator may be needed.
People with SMA also need to be watched for choking. This is because the muscles that control swallowing are weak.
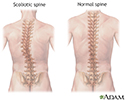
Physical therapy is important to prevent contractions of muscles and tendons and abnormal curvature of the spine (scoliosis). Bracing may be needed. Surgery may be needed to correct skeletal deformities, such as scoliosis.

Outlook (Prognosis)
Without treatment, children with SMA type I rarely live longer than 2 to 3 years because of respiratory problems and infections. Survival time with type II is longer, but the disease kills most of those who are affected while they are still children. New therapies that increase the amount of SMN protein have resulted in infants and children living much longer.
Children with type III disease may survive into early adulthood. But, people with all forms of the disease have weakness and disability that gets worse over time. Adults who develop SMA often have a normal life expectancy.
Possible Complications
Complications that may result from SMA include:
- Aspiration (food and fluids get into the lungs, causing pneumonia)
- Contractions of muscles and tendons
- Heart failure
- Scoliosis

When to Contact a Medical Professional
Contact your provider if your child:
- Appears weak
- Develops any other symptoms of SMA
- Has difficulty feeding
Breathing difficulty can rapidly become an emergency condition.

Prevention
Genetic counseling is recommended for people with a family history of SMA who want to have children.

References
Fearon C, Murray B, Mitsumoto H. Disorders of upper and lower motor neurons. In: Jankovic J, Mazziotta JC, Pomeroy SL, Newman NJ, eds. Bradley and Daroff's Neurology in Clinical Practice. 8th ed. Philadelphia, PA: Elsevier; 2022:chap 97.
Manzur AY. Evaluation and investigation of neuromuscular disorders. In: Kliegman RM, St. Geme JW, Blum NJ, Shah SS, Tasker RC, Wilson KM, eds. Nelson Textbook of Pediatrics. 22nd ed. Philadelphia, PA: Elsevier; 2025:chap 647.
NIH-National Institute of Neurological Disorders and Stroke website. Spinal muscular atrophy. www.ninds.nih.gov/health-information/disorders/spinal-muscular-atrophy. Updated November 28, 2023. Accessed March 14, 2024.
Review Date: 12/31/2023

 All rights reserved.
All rights reserved.